Il nuovo regolamento MDR 2017/745 è un cambiamento importante rispetto alla 93/42, i laboratori odontotecnici dopo 20 anni hanno dovuto rivedere la modulistica prodotta ed implementare le specifiche indicazioni del nuovo regolamento.
A differenza della normativa 93/42 dove era ben chiaro i compiti dei fabbricanti di dispositivi su misura al fine di redarre la dichiarazione di conformità, con il nuovo regolamento troviamo le procedure richieste al fabbricante di dispositivi su misura solo all’allegato XIII, mentre è più complicato ricercare all’interno del regolamento, che riguarda la fabbricazione di dispositivi medici in generale, le parti che devono essere applicate da coloro che realizzano dispositivi su misura.
Il principio generale è la classificazione dei dispositivi medici in classi a seconda della loro pericolosità, da ciò deriva l’applicazione degli adempimenti richiesti dal nuovo regolamento in modo diverso a seconda del dispositivo prodotto.
Il modus operandi dell’ odontotecnico con il nuovo regolamento non è cambiato in sostanza rispetto alla 93/42, i nuovi adempimenti vanno ad aggiungersi a quelli precedenti.
I software gestionali già presenti da anni nel settore, hanno implementato in modo diverso l’applicazione del regolamento MDR 2017/745, questo è dovuto al fatto che non c è una distinzione netta tra gli adempimenti del fabbricante su misura e non, l’unica distinzione fondamentale è quella derivante dalla classificazione di pericolosità del dispositivo.
I software per laboratorio odontotecnico per adeguarsi alla nuova normativa, oltre all’applicazione delle normative già presenti nella 93/42 come la gestione dei cicli di fabbricazione e il relativo fascicolo tecnico, dovrà prevedere una gestione del dato clinico, ovvero i dati relativi alle performance che il dispositivo a prodotto nel suo ciclo di vita.
I dati clinici sono ad esempio, la presenza di rotture o anomalie durante il ciclo di vita del dispositivo, un confort non adeguato per il paziente o un usura anomala.
I dati clinici si ottengono registrando in un database le riparazioni effettuate su un determinato dispositivo e tramite un sistema di sorveglianza post vendita che coinvolga il medico o la struttura sanitaria durante il ciclo di vita del prodotto.
Il dato clinico si può ottenere anche dalla letteratura, dalle riviste di settore, dalle banche dati pubbliche che raccolgano ricerche a riguardo del dispositivo, dalla ricerca sponsorizzata, dalle pubblicazioni scientifiche.
Il software gestionale deve avere una sezione per la raccolta di tali dati.
Un altro fattore non trascurabile che viene implementato nel software è l’analisi dei rischi standard precompilata che deve essere tenuta costantemente aggiornata. Il fabbricante non ha l’obbligo di fare una analisi dei rischi per ogni dispositivo su misura ma ha la possibilità di tenere una analisi dei rischi standard e ad ogni dispositivo poter dichiarare che è conforme a quell’analisi dei rischi.
I piani di fabbricazione già presenti nella normativa 93/42 dovranno essere aggiornati alle nuove tecnologie che prevedono progetti in digitale, dispositivi fresati con macchine a controllo numerico e stampaggio.
Il software gestionale dovrebbe avere già inserite le fasi di fabbricazione coerenti alle nuove tecnologie che andranno poi adattate ai processi di fabbricazione del laboratorio.
Il regolamento MDR 2017/745 prevede anche la nomina di un responsabile del regolamento 2017/745, il software deve prevedere la lettera di incarico del responsabile.
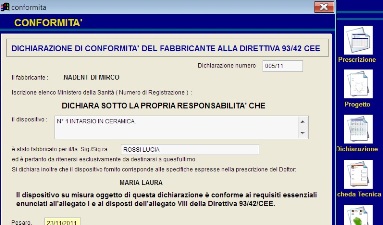
La direttiva prevede una serie di moduli che il software Primolab può produrre: tra le varie stampe richieste le principali sono:
Dichiarazione di Conformità
Non conformità (nel caso alcuni dei controlli siano falliti)
Progettazione
Istruzioni d’uso
Scheda tecnica con lotti impiegati
Etichetta con nome completo fabbricante
Scheda rischi e controlli
Attestato di consegna
Scheda di sorveglianza Post-vendita
Il software Primolab in modalita stampa fronte retro automatica (per le stampanti predisposte), può stampare tutti i documenti da consegnare al paziente in solo foglio ripiegabile a metà con quattro facciate.